El Observatorio de Salud y Medicina Integrativa ha podido tener acceso a este estudio del investigador Heiko Braak y demás compañeros, cuya hipótesis es que el origen del Parkinson está en el intestino. A continuación, te lo mostramos en español. ¡Comienza a leer! ⬇️
Resumen
La enfermedad de Parkinson esporádica involucra múltiples sistemas neuronales y es el resultado de cambios que se desarrollan en algunos tipos de células nerviosas susceptibles, esenciales para el diagnóstico neuropatológico: son las neuritas de Lewy y los cuerpos de Lewy inmunopositivos para α-sinucleína.
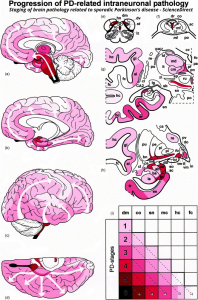
El proceso patológico se dirige a sitios de inducción específicos: las lesiones ocurren inicialmente en el núcleo motor dorsal de los nervios glosofaríngeo y vago y el núcleo olfatorio anterior. A partir de entonces, los grises nucleares y las áreas corticales menos vulnerables se ven afectados gradualmente. El proceso patológico en el tronco encefálico sigue un curso ascendente con poca variación interindividual. La patología en el núcleo olfatorio anterior incursiona menos en áreas relacionadas que la que se desarrolla en el tronco encefálico. Se produce afectación cortical, comenzando con la mesocorteza temporal anteromedial. A partir de ahí, la neocorteza sucumbe, comenzando con la asociación sensorial de alto orden y las áreas prefrontales. Las áreas de asociación sensorial/premotoras de primer orden y los campos sensoriales/motores primarios siguen su ejemplo.
Este estudio rastrea el curso de la patología en casos de Parkinson incidentales y sintomáticos y propone un procedimiento de estadificación basado en la extensión topográfica fácilmente reconocible de las lesiones.
1. Introducción [resumen]
La enfermedad de Parkinson (EP) esporádica es una enfermedad degenerativa progresiva del sistema nervioso humano que se manifiesta clínicamente después de que la patología ya ha alcanzado una etapa avanzada. Un requisito previo para el diagnóstico post-mortem de las fases presintomática y sintomática del proceso patológico que subyace a la EP es la evidencia de cuerpos de inclusión específicos que se desarrollan como neuritas de Lewy (LN) fusiformes o filiformes en los procesos celulares, y en la forma de cuerpos de Lewy globulares (LB) en el pericario neuronal. En la esporádica, solo unos pocos tipos específicos de células nerviosas son propensas a desarrollar las lesiones.
Un componente principal de los LN y los LB es una forma agregada de la proteína α-sinucleína normalmente presináptica. Todavía se desconoce por qué esta proteína hidrófila deja sus sitios de unión dentro de los botones sinápticos y, junto con otros componentes como los neurofilamentos fosforilados y la ubiquitina, una proteína de choque térmico requerida para la descomposición de proteínas anormales no dependientes de ATP lisosomal, se transforma gradualmente en virtualmente LN o LB insolubles. Surge la pregunta de si la patología evoluciona simultáneamente en todos estos sitios de inducción nigral y extranigral o si los diversos sitios difieren en sus susceptibilidades para desarrollar las alteraciones relacionadas con la enfermedad y, en consecuencia, siguen una secuencia coherente.
El presente estudio, por lo tanto, incluye intencionalmente un espectro de casos que exhiben LN y LB en un subconjunto específico de tipos neuronales y sitios de predilección, que se sabe que están involucrados en casos clínicos de EP. Al hacerlo, se asumime que es correcto que los casos asintomáticos y sintomáticos se pueden ordenar de tal manera que los casos que presentan la patología más leve representan el punto de partida y los más afectados el término de un espectro de enfermedad, con una tendencia a aumentar la gravedad. por parte de la patología global. De acuerdo con esta suposición, el daño neuronal no se desarrolla al azar, sino que sigue una secuencia predeterminada marcada por cambios característicos en la extensión topográfica.
Esta investigación tiene como objetivo elaborar un procedimiento de estadificación neuropatológica basado en la topografía de estos cambios. Se destaca que la muestra del estudio no incluye casos clínicamente diagnosticados como enfermedad de LB difuso.
2. Metodología [resumen]
Se estudiaron tres grupos de casos. El primero consistió en cerebros obtenidos en la autopsia de 41 individuos con diagnóstico clínico de EP (19 mujeres, 22 hombres, edad 75,7 ± 7,2 años). Los protocolos clínicos de estos casos señalaron el predominio del temblor o la rigidez combinados con hipocinesia e inestabilidad postural. El tejido cerebral exhibió LB nigral y pérdida severa de neuronas cargadas de neuromelanina nigral.
El segundo incluyó cerebros de autopsia de 69 individuos. En la mayoría de estos, la historia clínica no hacía referencia a los síntomas asociados a la EP. Unos pocos casos con patología severa fueron mal diagnosticados o no recibieron un diagnóstico clínico. Los 69 casos mostraron la presencia de LN y/o LB en un subconjunto de tipos neuronales en los sitios de predilección antes mencionados (35 mujeres, 34 hombres, edad 76,1 ± 7,9 años).
El tercer grupo, que incluía 58 casos emparejados por edad y sexo (25 mujeres, 33 hombres, 75,9 ± 8,2 años de edad) se utilizó para la comparación. Las historias clínicas previas de estos casos no incluían antecedentes de enfermedad neurológica o psiquiátrica. Ninguno de estos casos contenía LB/LN en el núcleo motor dorsal IX/X. Para la orientación topográfica, las secciones se tiñeron con pigmento de lipofuscina (aldehído-fucsina) así como con material de Nissl (rojo Darrow). Se empleó la tinción con aldehído-fucsina porque las propiedades de la pigmentación se pueden utilizar para distinguir los diferentes tipos de células nerviosas en el cerebro del adulto humano.
3. Resultados [resumen]
Los cerebros de todos los casos incidentales y de individuos con EP esporádica clínicamente manifiesta muestran la presencia de LN y LB inmunorreactivos a la sinucleína α mientras que al mismo tiempo están libres de inclusiones intracitoplasmáticas relacionadas con sinucleinopatías α no relacionadas con la EP. La patología concomitante relacionada con la EA se encuentra dentro del rango esperado de los respectivos grupos de edad.
Los LB suelen estar presentes como cuerpos de inclusión esféricos o reniformes, débilmente acidófilos, con superficies lisas, que varían en forma y tamaño. Los LB individuales o grupos de ellos normalmente se encuentran dentro de los depósitos de gránulos de lipofuscina o neuromelanina de las células nerviosas involucradas y normalmente no se ven entre los parches de sustancia de Nissl. En ocasiones aparecen “cuerpos pálidos” mal definidos y débilmente inmunopositivos entre el depósito de pigmento y el núcleo celular o adyacentes a una LB. Los LN gruesos tienen forma de maza o de sacacorchos. Otros son cortos y rechonchos o largos y filiformes. Los LN pueden adquirir una apariencia varicosa, y tanto los tipos de LN con forma de hilo como los más voluminosos tienden a bifurcarse repetidamente, a menudo terminando en agrandamientos en forma de lágrima.
La gravedad de la patología relacionada con la EP varía entre los casos y varía desde un solo LN (+) en el núcleo motor dorsal IX/X hasta densidades extremadamente altas de cuerpos de inclusión en múltiples sitios, incluida la corteza cerebral. La destrucción relacionada con la enfermedad se centra en distintos tipos neuronales dentro de grises nucleares particulares, así como en áreas y capas corticales específicas. Los tipos de células nerviosas susceptibles muestran diferencias sistemáticas con respecto al grado en que tienden a desarrollar LN/LB.
4. Discusión [resumen]
Los cuerpos de inclusión que contienen α-sinucleína que se encuentran en los casos incidentales y sintomáticos son patológicos y, por lo general, no se consideran concomitantes normales del envejecimiento cerebral. Por su perfil inmunocitoquímico, difícilmente pueden confundirse con las lesiones que se dan en otras enfermedades neurodegenerativas que no pertenecen al grupo de las sinucleinopatías. Los sitios de inducción y los tipos de células implicados en la atrofia multisistémica o la enfermedad de Hallervorden-Spatz difieren de los afectados en la EP. En consecuencia, los cuerpos de inclusión aquí considerados se consideran indicativos de casos incidentales y sintomáticos de EP esporádica.
Los hallazgos del presente estudio corroboran la suposición de que las lesiones clave en la EP comienzan a desarrollarse, como en otras enfermedades neurodegenerativas, un tiempo considerable antes de la aparición de las disfunciones somatomotoras. Los LN y LB de casos incidentales muestran las mismas características morfológicas y perfiles inmunocitoquímicos, y se desarrollan notablemente en los mismos subconjuntos de células nerviosas y en los mismos sitios de predilección que los de los casos sintomáticos. El patrón lesional observado en los casos incidentales se complementa perfectamente con el presentado en los casos de EP completamente desarrollada. Solo la extensión topográfica limitada y el grado comparablemente leve de las lesiones distinguen los casos incidentales de aquellos con EP clínicamente manifiesta.
Se requiere un procedimiento de estadificación neuropatológica que permita una diferenciación suficiente entre las etapas inicial, intermedia y final de las lesiones relacionadas con la EP. Además, dicho procedimiento permitiría la identificación precisa de controles verdaderamente desprovistos de lesiones relacionadas con la EP. Dado que los cerebros de los ancianos con frecuencia se ven afectados por más de una enfermedad neurodegenerativa, una gran proporción de pacientes con EP presenta patología relacionada con la EA concurrente y viceversa. Por lo tanto, una evaluación post-mortem precisa de tales individuos requiere el uso de sistemas de estadificación post-mortem tanto para la EA como para la PD.